題目連結:https://rosalind.info/problems/orf/
今天這一題非常精彩,也是一連串實戰演練的流程
是以前這三篇作法的延續題目
開放閱讀框ORF,是指在一段mRNA序列中
從起始密碼子(Start codon)開始,到終止密碼子(Stop codon, TAA/TAG/TGA) 結束
中間沒有出現任何其他終止密碼子的區段
輸入:
>Rosalind_99
AGCCATGTAGCTAACTCAGGTTACATGGGGATGACCCCGCGACTTGGATTAGAGTCTCTTTTGGAATAAGCCTGAATGATCCGAGTAGCATCTCAG
輸出:
MLLGSFRLIPKETLIQVAGSSPCNLS
M
MGMTPRLGLESLLE
MTPRLGLESLLE
題目輸入一段DNA序列,要輸出可能出現的胺基酸序列
胺基酸序列可能存在多種,按照任意順序返回即可
流程如下:
DNA(A、T、C、G) -> RNA(A、U、C、G) -> 密碼子(三個核苷酸為一組) -> 胺基酸(20種英文字母)
理論上,蛋白質的合成是根據 mRNA 上的密碼子進行的
但在實務上,我們通常都使用DNA序列來搜尋ORF
為什麼?因為可以少一層「T <-> U」的轉換,以及文章尾的生物知識補充
所以我們這邊,不是使用RNA(A、U、C、G),而是DNA(A、T、C、G)
| 階段 | 生物學理論 | 生物資訊實作 |
|---|---|---|
| 轉錄與翻譯對象 | mRNA(A、U、C、G) | DNA(A、T、C、G) |
| 起始 codon | AUG | ATG |
| 終止 codon | UAA / UAG / UGA | TAA / TAG / TGA |
| 目的 | 真實的蛋白合成 | 找出潛在的基因區段(ORF) |
DNA_CODON_TABLE = {
"TTT":"F","TTC":"F","TTA":"L","TTG":"L",
"CTT":"L","CTC":"L","CTA":"L","CTG":"L",
"ATT":"I","ATC":"I","ATA":"I","ATG":"M",
"GTT":"V","GTC":"V","GTA":"V","GTG":"V",
"TCT":"S","TCC":"S","TCA":"S","TCG":"S",
"CCT":"P","CCC":"P","CCA":"P","CCG":"P",
"ACT":"T","ACC":"T","ACA":"T","ACG":"T",
"GCT":"A","GCC":"A","GCA":"A","GCG":"A",
"TAT":"Y","TAC":"Y","TAA":"*","TAG":"*",
"CAT":"H","CAC":"H","CAA":"Q","CAG":"Q",
"AAT":"N","AAC":"N","AAA":"K","AAG":"K",
"GAT":"D","GAC":"D","GAA":"E","GAG":"E",
"TGT":"C","TGC":"C","TGA":"*","TGG":"W",
"CGT":"R","CGC":"R","CGA":"R","CGG":"R",
"AGT":"S","AGC":"S","AGA":"R","AGG":"R",
"GGT":"G","GGC":"G","GGA":"G","GGG":"G"
}
STOP = {"TAA","TAG","TGA"}
def parse_fasta(data: str) -> dict:
sequences = {}
label = ""
for line in data.strip().splitlines():
line = line.strip()
if not line:
continue
if line.startswith(">"):
label = line[1:].strip()
if not label:
label = ""
continue
sequences[label] = ""
elif label:
sequences[label] += line
return sequences
def revcomp(dna: str) -> str:
comp = str.maketrans("ACGTacgt", "TGCAtgca")
return dna.translate(comp)[::-1]
def proteins_from_strand(dna: str) -> set:
n = len(dna)
out = set()
for i in range(n - 2):
if dna[i:i+3] == "ATG":
aa = []
for j in range(i, n-2, 3):
codon = dna[j:j+3]
aa_code = DNA_CODON_TABLE.get(codon)
if not aa_code:
break
if aa_code == "*": # stop codon
if aa:
out.add(''.join(aa))
break
aa.append(aa_code)
return out
def all_orf_proteins(fasta_text: str) -> dict:
fasta_dict = parse_fasta(fasta_text)
results = {}
for label, seq in fasta_dict.items():
seq = seq.upper()
rc = revcomp(seq)
proteins = set()
proteins |= proteins_from_strand(seq)
proteins |= proteins_from_strand(rc)
results[label] = proteins
return results
data = """
>Rosalind_99
AGCCATGTAGCTAACTCAGGTTACATGGGGATGACCCCGCGACTTGGATTAGAGTCTCTTTTGGAATAAGCCTGAATGATCCGAGTAGCATCTCAG
"""
res = all_orf_proteins(data)
for label, prots in res.items():
for p in prots:
print(p)
要先執行 pip install biopython 套件安裝
from Bio.Seq import Seq
dna = Seq("AGCCATGTAGCTAACTCAGGTTACATGGGGATGACCCCGCGACTTGGATTAGAGTCTCTTTTGGAATAAGCCTGAATGATCCGAGTAGCATCTCAG")
STOPS = {"TAA", "TAG", "TGA"}
def find_orfs(seq: Seq):
prots = set()
s = str(seq).upper()
n = len(s)
for frame in range(3):
i = frame
while i <= n - 3:
codon = s[i:i+3]
if codon == "ATG":
j = i + 3
found_stop = False
while j <= n - 3:
if s[j:j+3] in STOPS:
found_stop = True
break
j += 3
if found_stop:
prot = str(Seq(s[i:j]).translate())
prots.add(prot)
i += 3
return prots
proteins = set()
proteins |= find_orfs(dna)
proteins |= find_orfs(dna.reverse_complement())
for p in proteins:
print(p)
前面提到
實務上通常直接以DNA序列進行ORF搜尋
但嚴格上來講,不應該是這樣的...?
這與 RNA加工(轉錄後修飾)有關
只有真核生物會進行RNA加工,原核生物不會
所以原核生物用DNA來進行ORF搜尋完全沒問題、合情合理,因為原核基因沒有內含子(intron)
但是真核生物用DNA來搜尋會出事
真核的DNA經過轉錄之後,還會被加工才會變成真正的mRNA
在真核細胞中,完整流程:
DNA -> pre-mRNA(初級轉錄本) -> 成熟mRNA -> 離開細胞核 -> 到核糖體轉譯成胺基酸鏈其中,在 pre-mRNA(初級轉錄本) -> 成熟mRNA 這個步驟發生了RNA加工(RNA processing),做的事情包含:
- 5′ Capping(加帽):一個倒接的7-甲基鳥苷(7-methylguanosine, 簡寫m⁷G)
- 剪切(splicing):去掉內含子intron,保留外顯子exons
- 3′ Polyadenylation(加尾):一大堆重複的AAAAA序列
一條成熟的mRNA有以下五個部位:
| 區段 | 是否轉譯 | 功能簡述 |
|---|---|---|
| 5′ cap | ❌ | 保護 RNA、協助起始 |
| 5′UTR | ❌ | 調控翻譯效率 |
| ORF/CDS | ✅ | 翻譯成蛋白質 |
| 3′UTR | ❌ | 調控穩定性與終止 |
| poly-A tail | ❌ | 穩定 RNA、防止降解 |
但是,在生物資訊實務分析上
就算直接用mRNA序列,也要先去掉3' poly-A tail
因為在成熟的mRNA上,5' cap 跟 3' poly-A tail 都不會合成出胺基酸
真核細胞就是比較麻煩且複雜繁瑣...orz
| 項目 | 原核生物 | 真核生物 |
|---|---|---|
| 5′ cap | 無 | 有(m⁷G cap) |
| poly-A tail | 有,但作用為促進降解 | 有,作用為增加穩定性 |
| 5′UTR | 有(Shine–Dalgarno序列、riboswitch) | 有(Kozak序列) |
| 3′UTR | 有(短,調控降解) | 有(長,調控穩定性與轉譯效率) |
| 剪接(splicing) | 無 | 有(移除內含子) |
| 多基因轉錄(polycistronic) | 常見(含多個 ORF) | 多為單基因轉錄(monocistronic) |
哎,說來複雜拉

簡單來說
以前認為,DNA->RNA->蛋白質
這過程,就像寫一封信件請求或委託什麼
信件裡面(序列)寫啥,對方就會做啥(產生對應胺基酸)
委託寫啥,對方就產啥
但後來發現,哇這些分子怎麼跟人類社會一樣
信件一開始不是請求或委託
而是使用敬語、一些無關請求的客氣詞,套近乎,稱讚關心連連
就是5'UTR
真正要做的事情、委託寫在中間 CDS/ORF
尾部又是祝一切安好、身體健康吾皇萬歲等等,又與蛋白質無關,就是3'UTR
以前人類認為,只要手持一張胺基酸編碼表,就能打遍天下無敵手
後來發現編碼表只適用中間區段,前後的敬詞只是冗言贅字,與請求內容無關,完全無用、可以通通捨棄
但,這幾年又漸漸發現
不對欸!這些敬詞能讓人看得爽身心愉悅、或者身心俱疲乃至心情火大,有助於或者負面抑制於委託事項(胺基酸的產生)
於是正在研究這些敬詞到底是怎麼發揮調控作用的
說這麼多
我只是想說明,從DNA、或者mRNA序列,來直接分析取得ORF都不是100%準確
在真核生物上,用mRNA準確率會高許多
只不過,在實務上大家都使用DNA來做分析
加上基因庫資料幾乎都以DNA為主,算是一個歷史共業(?
好消息是
這一題練手題目不管這麼多,用DNA或RNA都可以就對了
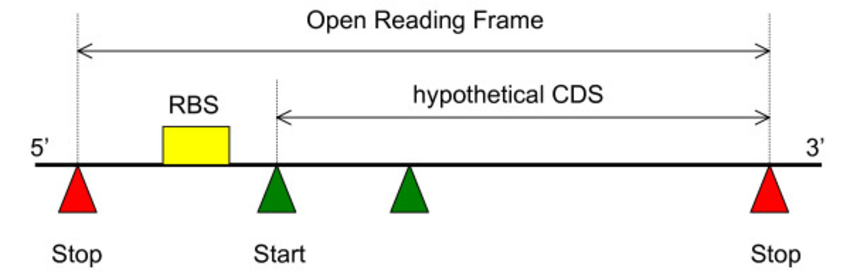
這兩個名詞相近、意思非常相似
ORF(Open Reading Frame) 是「理論上的可翻譯區段」
CDS(Coding Sequence) 是「實際被細胞轉譯成蛋白質的區段」
ORF: 電腦「看到」可能能翻譯的地方,所有排列組合
CDS: 生物「實際」用來翻譯蛋白的地方,實際上只會發生眾多排列組合中的某一種可能性。會依「生物狀態與環境條件」而改變
記憶方式:
ORF是候選區;CDS是經過驗證的區
ORF = Open潛在開放的
CDS = Confirmed確定會被翻譯的
生活比喻:
ORF:看了看錢包裡面剩餘的錢,理論上我今天晚餐可以去吃 八方雲集、六扇門、五花馬、四海遊龍、三商巧福、一蘭拉麵
CDS:但是到了吃飯時間,礙於其他家排隊人潮太多、或者今天特別想吃拉麵,所以我去吃了一蘭拉麵
某種程度上也算是一種,理想可能與現實方面的落差吧



 iThome鐵人賽
iThome鐵人賽